What is phylogeny?
Phylogeny refers to the representation of evolutionary relationships in family trees. It implies that all different organisms are genetically connected along the branches of a phylogenetic tree. When reading a phylogenetic tree, the roots of the tree represent the ancestral lineage and the tip of branches represent the descendants of the ancestor (Figure 1). A split in the lineage indicates speciation and is represented as branching in the tree. [1]
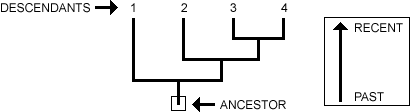
Figure 1. Root of the tree represents ancestor. Branches represent descendants. Moving from root to tips means moving forward from past to recent.
Species can be organized in clades when they evolve from a common ancestor. A clade shown in a phylogenetic tree is a grouping that includes a common ancestor and its descendants (Figure 2). [1]
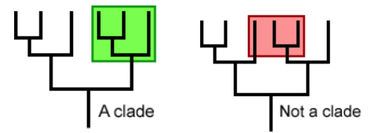
Figure 2. Clades are groupings that include common ancestor and its descendants.
Methods for Constructing Phylogenetic Trees
Maximum Likelihood Method (ML) and Neighbor Joining Method (NJ) are two major methods of phylogenetic tree reconstruction. NJ is a distance-based method. In this method, pairs that minimize the total branch length are found and branch lengths are obtained. The pair with the smallest length is the closest neighbor and is joined. The tree is reconstructed, and branch length is recalculated in the addition of new branches. This process repeats until one terminal is present. This method is fast and efficient. [2] The ML method provides probabilities of sequences using a model of their evolution where all possible trees are considered. It calculates the likelihood for each tree and chooses the one with the maximum likelihood. This method is the slower but most informative. [3]
Constructing Phylogenetic Trees
Identify the sequences of homologs
Use BLAST to gather sequences of homologs as FASTA text file formats. Other tools such as Homologene in the GeneBank can also be used to find the homologs’ sequences. Below is the FASTA file of the sequences of all SGSH homologs of interest.
Sequence alignment
Use ClustalOMEGA or MEGA programs to automatically align all the sequences. This will allow us to compare all the sequences. Below is a snapshot of a section of SGSH protein sequence alignment. The asterisks show the regions where the amino acids are conserved across the species. This can also be found by analyzing the colors used across species.
Tree construction
Use the aligned sequences and construct trees in ClustalOMEGA or MEGA.
Use BLAST to gather sequences of homologs as FASTA text file formats. Other tools such as Homologene in the GeneBank can also be used to find the homologs’ sequences. Below is the FASTA file of the sequences of all SGSH homologs of interest.
Sequence alignment
Use ClustalOMEGA or MEGA programs to automatically align all the sequences. This will allow us to compare all the sequences. Below is a snapshot of a section of SGSH protein sequence alignment. The asterisks show the regions where the amino acids are conserved across the species. This can also be found by analyzing the colors used across species.
Tree construction
Use the aligned sequences and construct trees in ClustalOMEGA or MEGA.
Maximum Likelihood Method (ML)
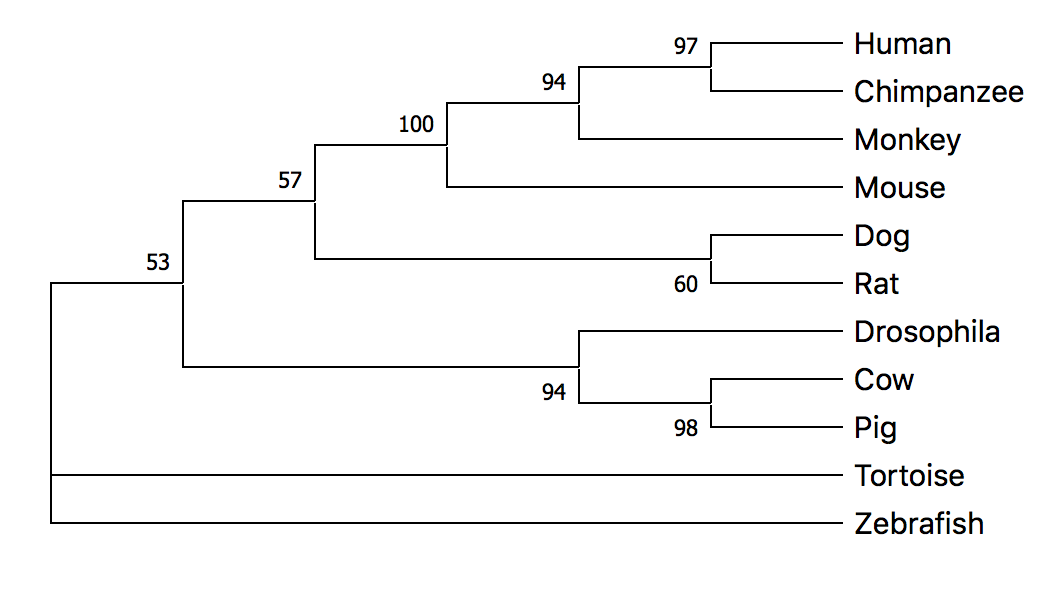
|
Neighbor Joining Method (NJ)
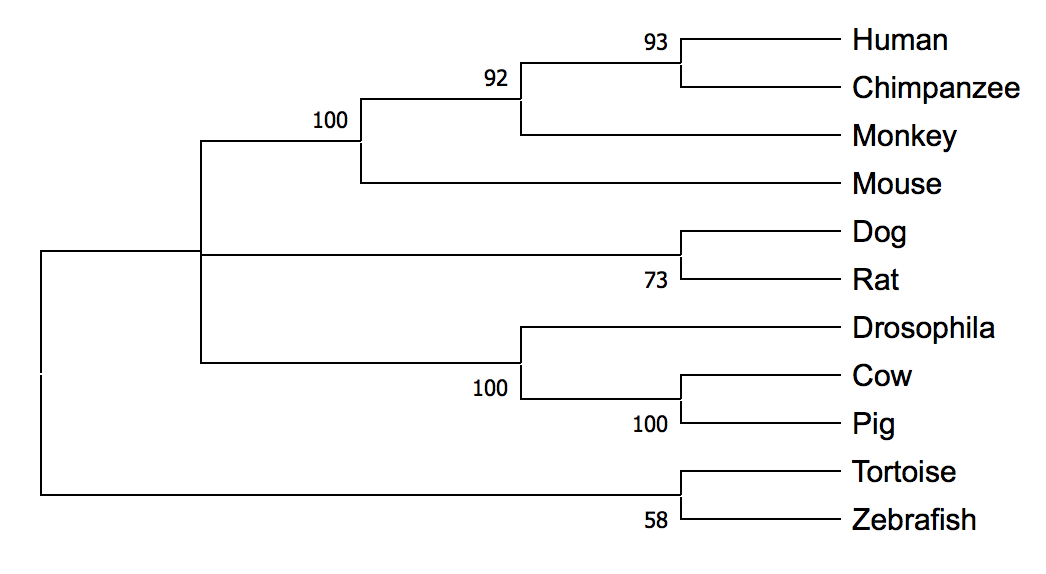
|
Discussion
Both the maximum likelihood and neighbor joining methods resulted in similar phylogenetic trees for SGSH homology sequences. The conservation of this protein over time is visible in the tree and confirms the importance of SGSH in neuronal function and survival of the species. Most species sharing more similarities and homology were found closer to each other in the trees. An exception to this observation was Drosophila which was in the same clade as cow and pig. This was a surprising finding and I plan to further investigate the Drosophila sequence.
References
[1] https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_05
[2] https://www.ncbi.nlm.nih.gov/pubmed/3447015
[3]https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
Header: https://britishlibrary.typepad.co.uk/.a/6a00d8341c464853ef017c373404fe970b-pi?_ga=2.216904022.197838224.1584072448-1168102271.1584072448
[2] https://www.ncbi.nlm.nih.gov/pubmed/3447015
[3]https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
Header: https://britishlibrary.typepad.co.uk/.a/6a00d8341c464853ef017c373404fe970b-pi?_ga=2.216904022.197838224.1584072448-1168102271.1584072448
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
