Introduction
Sanfilippo syndrome is a rare lysosomal storage disorder, affecting about 1 in a million people. This disease is an autosomal recessive disorder and primarily affects the function of lysosomes and lysosomal trafficking. In normal cells, lysosomes are essential organelles that function as the recyclers of the cells. They break down unwanted macromolecules and cellular waste using various enzymes. Sanfilippo syndrome type A, which is most common and most severe subtype, is caused by the accumulation of heparan sulfate GAGs in the lysosomes. Sanfilippo syndrome symptoms progress over time. Children affected don’t usually show any symptoms until early childhood, ages 2-6, where they start to lose their previously learned skills and show signs of developmental disability.
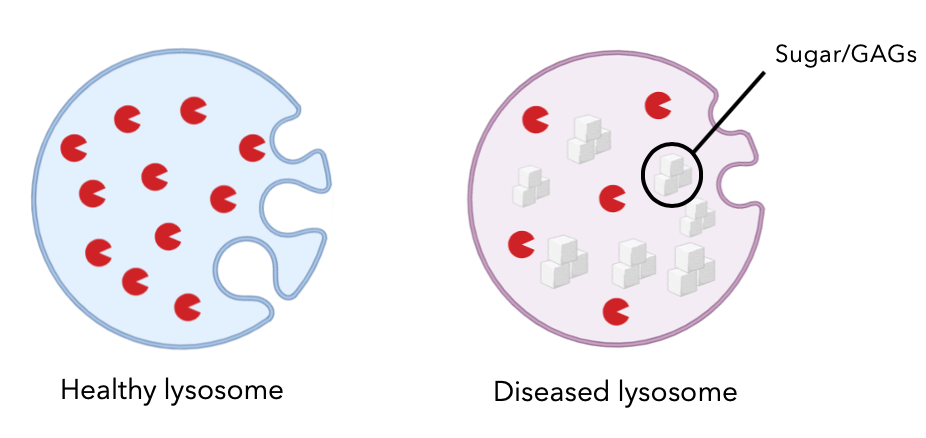
|
SGSH Gene
Sanfilippo Syndrome type A is caused by a mutation in the gene encoding N-sulfoglucosamine sulfohydrolase (SGSH). SGSH encodes enzyme sulfamidase which is involved in the breakdown of GAGs. This gene is primary localized in the lysosomes and involves biological process such as catabolism and brain development.
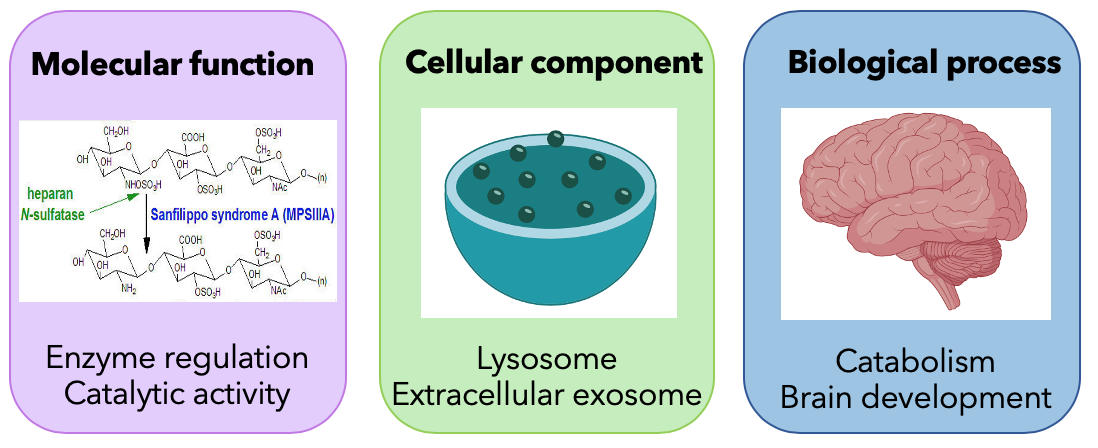
|
SGSH is also well conserved across species and model organisms. As shown in the phylogenetic tree, mice are the closest model organisms to human, which is one of the reasons I chose mice as my model organism.
SGSH has two domains that are also well conserved. Sulfatases are again enzymes involved in metabolic processes. And DUF is a domain of unknown function with no specific information. These domains were conserved in the model organisms.
SGSH has two domains that are also well conserved. Sulfatases are again enzymes involved in metabolic processes. And DUF is a domain of unknown function with no specific information. These domains were conserved in the model organisms.

|
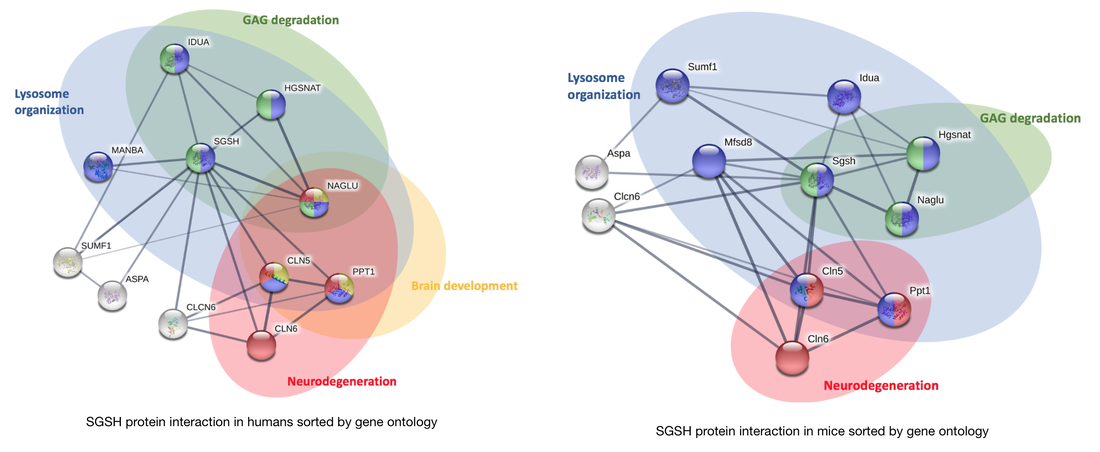
Looking at the protein interactions, you can see that there are some proteins involved in brain development and neurodegeneration that interact with SGSH. This interaction network reveals possible functions in brain development. This is similar in mice, where there is possible function in brain development.
Mice as model organisms |
I used mice as my model organism, because SGSH and the two domains were well conserved between human and mice. In addition, mice have the most similar brain complexity to humans in comparison to the other model organisms I found, which were not mammals. I was also interested in studying the lifespan since the symptoms progress over time. Knowing that one year in human is on average equal to nine days in mice, I can figure out the age in mice to study these critical points in life.
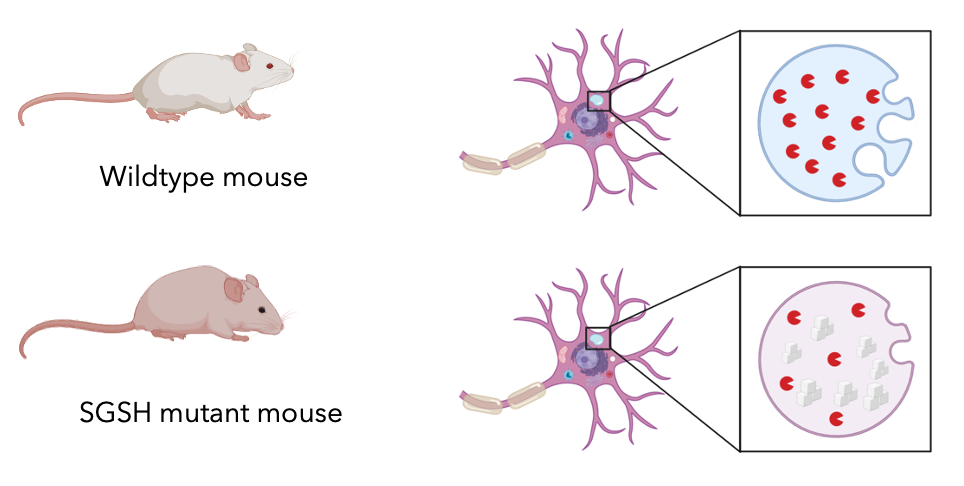
|
Gap in knowledge
Despite all the research in this area, it’s not clear why these sugar accumulations in the lysosome mostly affect the nervous system in Sanfilippo patients. Thus, my goal is to determine how sugar accumulates in the lysosomes of the brain. I hypothesize that sugar accumulation in lysosome is mediated by SGSH during early development.
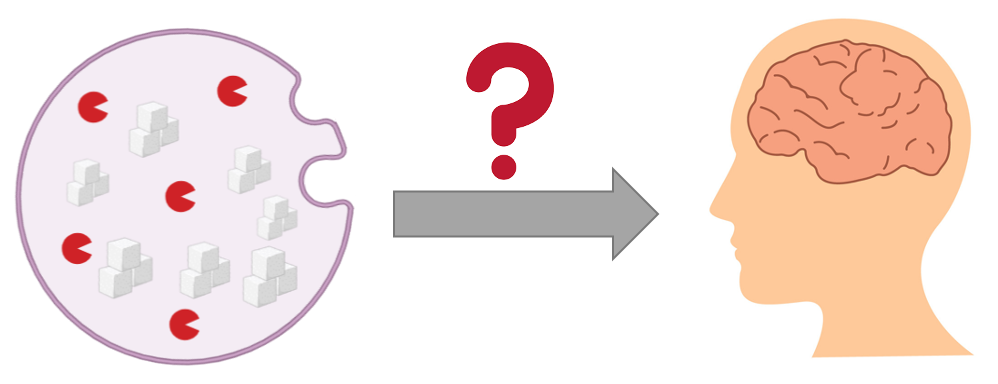
|
Aim1: Identify conserved amino acids in SGSH that are important for GAGs accumulation in neurons
My first aim is to identify conserved amino acids region of SGSH that may be important for removal of sugar from neurons over time. I will use BLAST to identify the homologs and then use ClustalOMEGA to align the sequences and then identify the regions where most amino acids are conserved. I chose the region where most serines were conserved within the sulfatase domain.
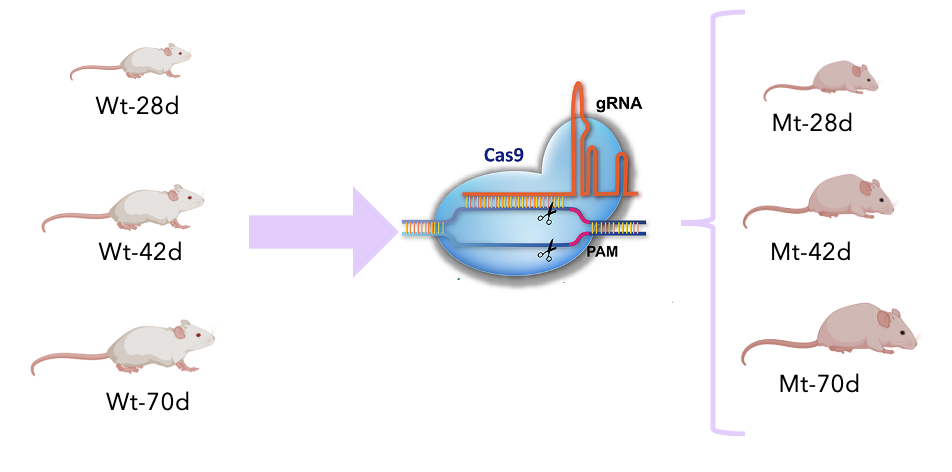
Then I would use CRISPR/Cas9 to create knockout mice at the three different ages, representing the 2, 6, and 15 year olds in human.
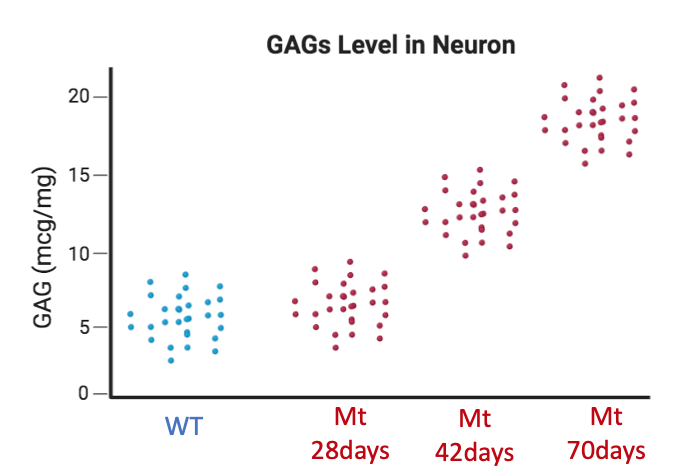
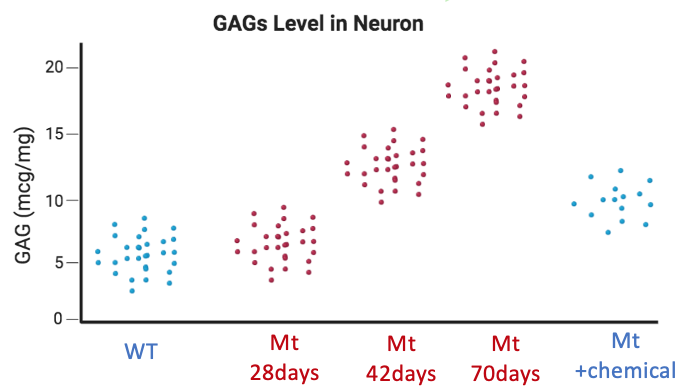
Lastly, I would develop a GAGs assay to quantify the level of sugar in the neurons over time. I hypothesize that sugar level increases with age in the mutant mice.
Then I would use CRISPR/Cas9 to create knockout mice at the three different ages, representing the 2, 6, and 15 year olds in human.
Lastly, I would develop a GAGs assay to quantify the level of sugar in the neurons over time. I hypothesize that sugar level increases with age in the mutant mice.

|
|
|
Aim 2: Identify small molecules that rescue neurodegeneration in SGSH mutants
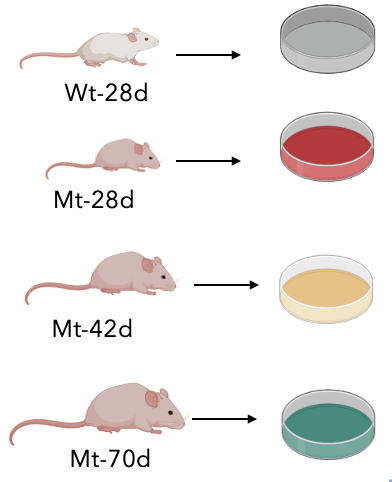
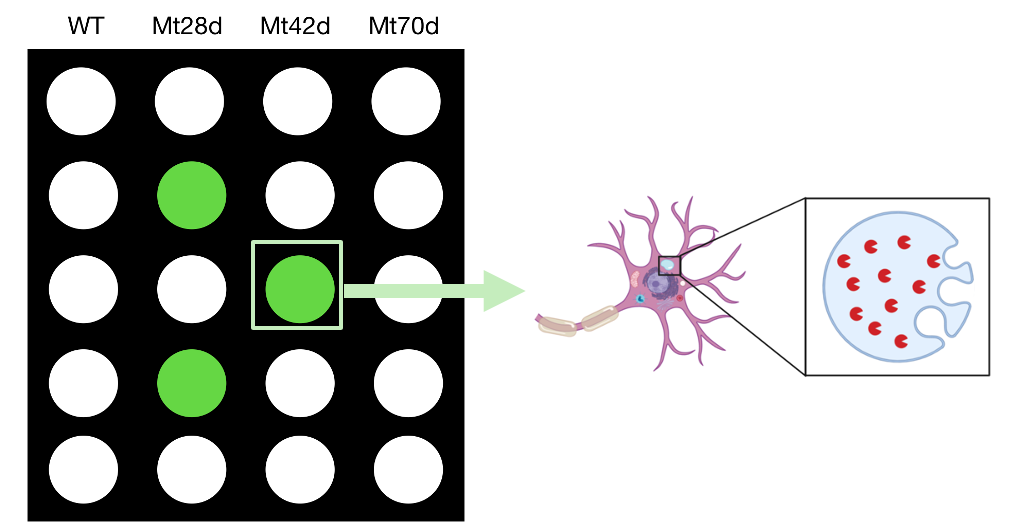
Moving onto the second aim, I wanted to identify small molecules that might rescue the phenotype of Sanfilippo syndrome. To do this, I would first prepare neuronal cell cultures using the mouse models. I would use a diverse chemical library and look for positive hits, that can rescue the phenotype in which the mutant at a late stage would show a normal lysosome without any accumulations.
The next goal is to determine what those molecules are and whether or not they can rescue sugar accumulation when you treat the mutant mice with that molecule. I hypothesize there will be molecule that would decrease the GAGs level in mutant mice.
The next goal is to determine what those molecules are and whether or not they can rescue sugar accumulation when you treat the mutant mice with that molecule. I hypothesize there will be molecule that would decrease the GAGs level in mutant mice.
|
|
|
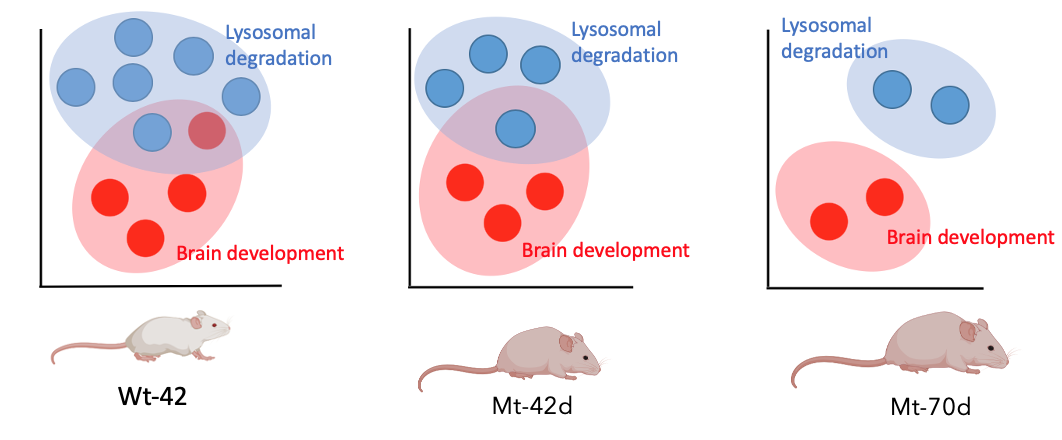
Aim 3: Quantify proteins associated with neurodegeneration and lysosomal degradation
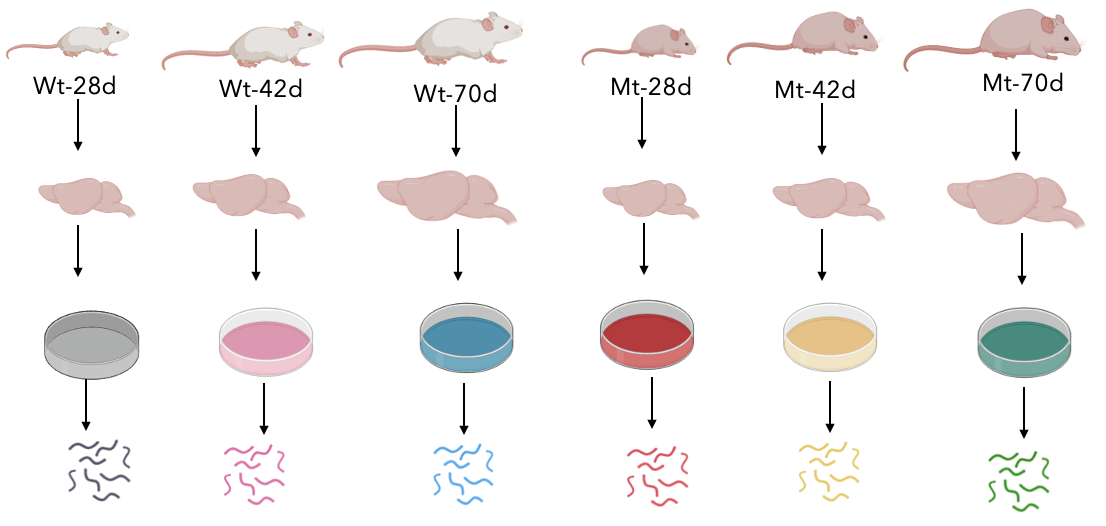
My last aim is to identify other proteins that are associated with brain development and interact with SGSH. I would first isolate brain tissues from both the wildtype and mutant models at the three different times, extract the proteins, and perform enzyme digestion to prepare the samples for iTRAQ.
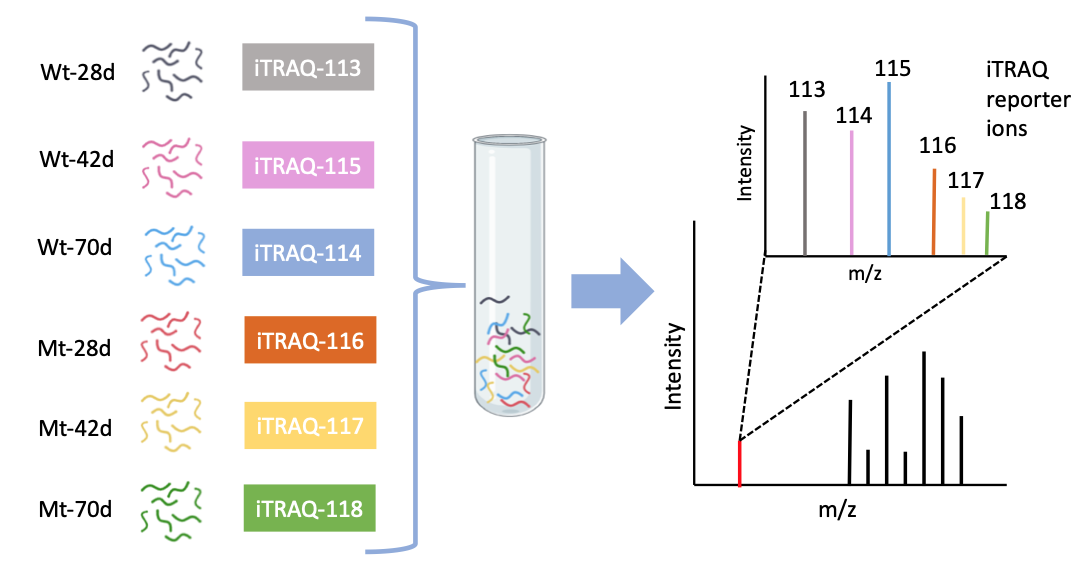
The peptides would then be labeled with iTRAQ tags, and then the samples were mixed. Then the results can be quantified using mass spec where the intensity of each iTRAQ ion is shown here.
Lastly, I would categorize the proteins using gene ontology and I hypothesize that the proteins involved in brain development and lysosomal degradation would be missing or decreased in the mutant mice with age.
The peptides would then be labeled with iTRAQ tags, and then the samples were mixed. Then the results can be quantified using mass spec where the intensity of each iTRAQ ion is shown here.
Lastly, I would categorize the proteins using gene ontology and I hypothesize that the proteins involved in brain development and lysosomal degradation would be missing or decreased in the mutant mice with age.
|
|
|
Future Directions
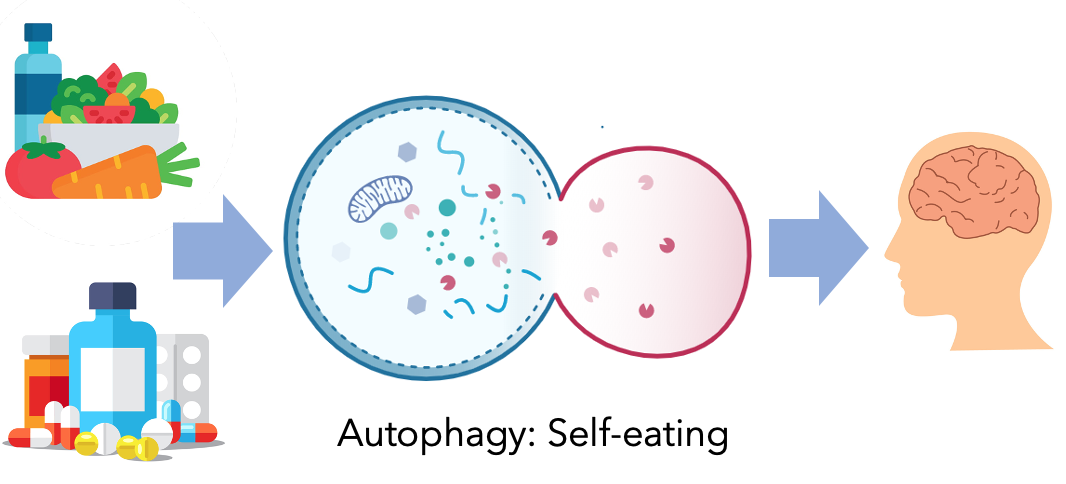
As for future directions, it would be really interesting to look into the process of autophagy and how it relates to Sanfilippo syndrome. Autophagy is a mechanism where the cell removes waste and it can be induced by certain drugs or diet. So I would be interested to see if a change in diet or using autophagy enhancers could potentially affect the phenotype in Sanfilippo syndrome.
|
| pourdashti_2_27_20.pptx |
| pourdashti_4_5_20.pdf |
| pourdashti_4_23_20.pdf |
References
Cure Sanfilippo Syndrome Foundation. Retrieved from: https://curesanfilippofoundation.org/what-is-sanfilippo/
Fedele A. O. (2015). Sanfilippo syndrome: causes, consequences, and treatments. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4664539/
Gilkes JA, Heldermon CD. Mucopolysaccharidosis III (Sanfilippo Syndrome)- disease presentation and experimental therapies. (2014). Retrieved from: https://www.ncbi.nlm.nih.gov/pubmed/25345095
National MPS Society. Retrieved from: https://mpssociety.org/learn/diseases/mps-iii/
de Ruijter, J. (2013). Sanfilippo disease (mucopolysaccharidosis type III): Early diagnosis and treatment. Retrieved from: https://pure.uva.nl/ws/files/2234091/126751_thesis.pdf
Pena, A. (2018). Advances in Substrate Reduction Therapy Show Promise for Sanfilippo Syndrome. Retrieved from: https://sanfilipponews.com/2018/05/15/advances-substrate-reduction-therapy-show-promise-sanfilippo-syndrome/
Images:
Header: gene-header.jpg
All images are from www.Biorender.com
Fedele A. O. (2015). Sanfilippo syndrome: causes, consequences, and treatments. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4664539/
Gilkes JA, Heldermon CD. Mucopolysaccharidosis III (Sanfilippo Syndrome)- disease presentation and experimental therapies. (2014). Retrieved from: https://www.ncbi.nlm.nih.gov/pubmed/25345095
National MPS Society. Retrieved from: https://mpssociety.org/learn/diseases/mps-iii/
de Ruijter, J. (2013). Sanfilippo disease (mucopolysaccharidosis type III): Early diagnosis and treatment. Retrieved from: https://pure.uva.nl/ws/files/2234091/126751_thesis.pdf
Pena, A. (2018). Advances in Substrate Reduction Therapy Show Promise for Sanfilippo Syndrome. Retrieved from: https://sanfilipponews.com/2018/05/15/advances-substrate-reduction-therapy-show-promise-sanfilippo-syndrome/
Images:
Header: gene-header.jpg
All images are from www.Biorender.com
Sheida Pourdashti
[email protected]
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Last updated: 5/1/2020
[email protected]
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Last updated: 5/1/2020

{kind=link}